Résumé
Les tumeurs hypophysaires sont bénignes mais causent des problèmes importants à la fois par leurs effets compressifs sur les structures cérébrales et par les syndromes d’excès ou de déficits hormonaux qu’elles peuvent engendrer. La thérapie endocrinienne est devenue de plus en plus efficace, avec le développement d’agonistes de la dopamine et d’analogues de la somatostatine. Récemment des antagonistes du récepteur de l’hormone de croissance ont été développés par modification de la molécule de GH. Il est vraisemblable que des avancées futures basées sur notre compréhension de la biologie vont améliorer profondément le traitement de ces tumeurs
Petites, bénignes et quelquefois inquiétantes
Les tumeurs hypophysaires sont étonnement fréquentes-environ 20% d’entre nous vont développer un petit adénome dans l’hypophyse au cours de notre vie. Chez la plupart elles ne seront pas détectées, car elles sont généralement petites et ne sécrètent pas d’excès significatif d’aucune hormone hypophysaire. Ces micro-adénomes sont de plus en plus observés, incidemment, sur des tomodensitométries cérébrales. Dans une certaine proportion d’individus, cependant, les tumeurs hypophysaires secrètent une des six hormones hypophysaires en quantités suffisamment importantes pour causer un syndrome clinique. De nombreuses tumeurs ne secrètent pas beaucoup d’hormone fonctionnelle mais certaines deviennent suffisamment grosses pour comprimer les nerfs optiques dans leur chemin de l’œil au cerveau ; elles érodent l’ os environnant la glande ou compriment le reste du tissus hypophysaire normal. Ces problèmes cliniques- syndromes endocriniens, perte visuelle, migraines et hypopituitarisme- sont tout à faits rares, mais ils causent de sérieuses difficultés à leurs porteurs et peuvent causer la mort prématurée.
La biologie des tumeurs hypophysaires a largement évolué depuis les 20 dernières années et il est maintenant normal de considérer ces tumeurs en terme de leur type cellulaire d’origine. Les mesures des niveaux d’hormone circulante et l’analyse immunocytochimique d’échantillons de tumeurs ont permis de définir les catégories de tumeurs non fonctionnelles et de tumeurs fonctionnelles qui le plus souvent secrètent la prolactine, l’hormone de croissance ou l’ ACTH. Les syndromes cliniques respectifs de l’excès hormonal observé avec les prolactinomes, l’ acromégalie ou la maladie de Cushing sont bien caractérisés et le diagnostic peut être établi facilement dans la plupart des cas.
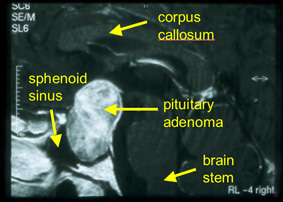
Image par résonance magnétique (tomodensitométrie) d’un volumineux adénome hypophysaire.
Chirurgiens et rayons X
Un traitement évident pour une tumeur bénigne gênante consiste à l’enlever. Atteindre l’hypophyse n’est pas facile et les techniques chirurgicales nécessitent l’accès au sinus sphénoïde à la base de la glande. La réussite demande le retrait de la tumeur entière, située dans un enfoncement profond inaccessible, sans endommager les structures normales. Réduire la masse peut être suffisant pour les tumeurs non fonctionnelles, mais les tumeurs endocrines sécrétantes doivent être éliminées en totalité pour assurer une guérison adéquate du syndrome clinique. Il n’est pas surprenant que même dans les mains les plus expertes, le taux de guérison endocrinienne est couramment de 80%-90% au mieux et peu descendre à 40%. Les tumeurs hypophysaires croissent très lentement sur plusieurs années, et la radiothérapie externe travaille aussi lentement, tuant les cellules qui occasionnellement essaient de se diviser. Ceci signifie que la radiothérapie est excellente pour prévenir la croissance de restes hypophysaires après une chirurgie partiellement réussie, mais ses effets sont lents et l’amélioration de l’hypersécrétion hormonale peut demander jusqu’ à 12-15 ans.
Applications ultérieures de la biologie
La thérapie endocrinienne des tumeurs hypophysaires a été découverte au début des années 70, quand les scientifiques de ” Sandoz Pharmaceuticals ” (maintenant ” Novartis “) ont découvert qu’une nouvelle drogue, le CB154, inhibait la sécrétion de la prolactine. Cette drogue, maintenant connue sous le nom de ” bromocryptine ” agit sur les récepteurs dopaminergiques D2 et était capable de normaliser le niveau de la prolactine chez des patients porteurs de prolactinomes. De façon encore plus impressionnante cette drogue induisait une rétraction dramatique du volume des tumeurs, même des plus grosses, évitant ainsi le besoin de chirurgie. Une série de drogues agonistes de la dopamine ont été développées ultérieurement, telles que la cabergoline et la quinagolide, et elles sont devenues les traitements préférés pour les patients avec ce type de tumeur.
Avec les succès étonnants des agonistes dopaminergiques, d’autres thérapies candidates ont été conçues. La somatostatine est une hormone peptidique distribuée largement dans le corps et impliquée dans la régulation de la sécrétion de nombreuses hormones. Son potentiel pour le traitement des tumeurs secrétant la GH a été reconnu très tôt mais son efficacité était limitée par sa très courte demi-vie et l’absence de spécificité pour GH. Pour surmonter ces problèmes des analogues de la somatostatine ont été synthétisés qui possèdent des demi-vies beaucoup plus longues et une spécificité plus grande pour les récepteurs de la somatostatine ( st2 et st5) concernés par la sécrétion de GH. Actuellement les analogues de la somatostatine constituent le traitement principal des tumeurs sécrétantes à GH et sont également utilisés pour les rares tumeurs sécrétantes à TSH. L’ octréotide et le lanréotide contrôlent la sécrétion de GH chez la majorité des patients acromégaliques et dans une minorité des cas ils causent une certaine rétraction de la tumeur.
Cependant, même avec les meilleurs traitements disponibles, la sécrétion de GH ne peut pas être contrôlée chez une minorité significative de patients. Pour résoudre ce problème une nouvelle classe de drogues ont été développées qui n’ont pas pour objectif de réduire le niveau de GH mais plutôt d’inhiber l’action de la GH. Le Pegvisomant est une protéine recombinante analogue de GH avec des substitutions d’acides aminés dans la région du site de liaison au récepteur, de sorte que la dimérisation du récepteur est empêchée et sa fonction bloquée. Le peptide a été conjugué au polyéthylène glycol afin de prolonger sa demi-vie jusqu’à 70 heures. Les études chez des patients acromégaliques suggèrent qu’il est efficace chez virtuellement tous les patients et on espère qu ‘il obtiendra une licence et sera plus largement disponible en 2003.
Thérapie génique
Les thérapies endocriniennes seront certainement raffinées dans les années à venir, mais il y a un intérêt récent pour la possibilité d’utiliser la thérapie génique pour traiter les tumeurs hypophysaires. La thérapie génique signifie simplement le transfert de gènes in vivo pour un bénéfice thérapeutique, et a été utilisée jusqu’à présent pour tenter le traitement de maladies fatales. Les applications actuelles incluent le remplacement d’enzymes absentes ou défectueuses ou d’autres protéines, ou bien l’ introduction de gènes ” suicides ” dont la protéine produite tue la cellule hôte. Cette dernière approche peut offrir quelque promesse pour les tumeurs hypophysaires agressives qui sont résistantes à toutes les thérapies standard. Les promoteurs spécifiques de gènes hypophysaires peuvent cibler des transgènes aux types cellulaires correspondants, et des études pré-cliniques évaluent différents virus ” vecteurs ” pour leur efficacité et leur sécurité. Ces applications deviennent rapidement faisables et les conséquences pour la sécurité étant mieux comprises, elles pourraient bien modifier les stratégies thérapeutiques pour les maladies hypophysaires dans l’avenir.
Traduction : Andrée Tixier-Vidal, UMR 7101 CNRS Université Pierre et Marie Curie, Paris
Cette brève est produite par la British Society for Neuroendocrinology et peut être utilisée librement pour l’enseignement de la neuroendocrinologie et la communication vers le public.
©British Society for Neuroendocrinology et Société de Neuroendocrinologie pour la traduction.